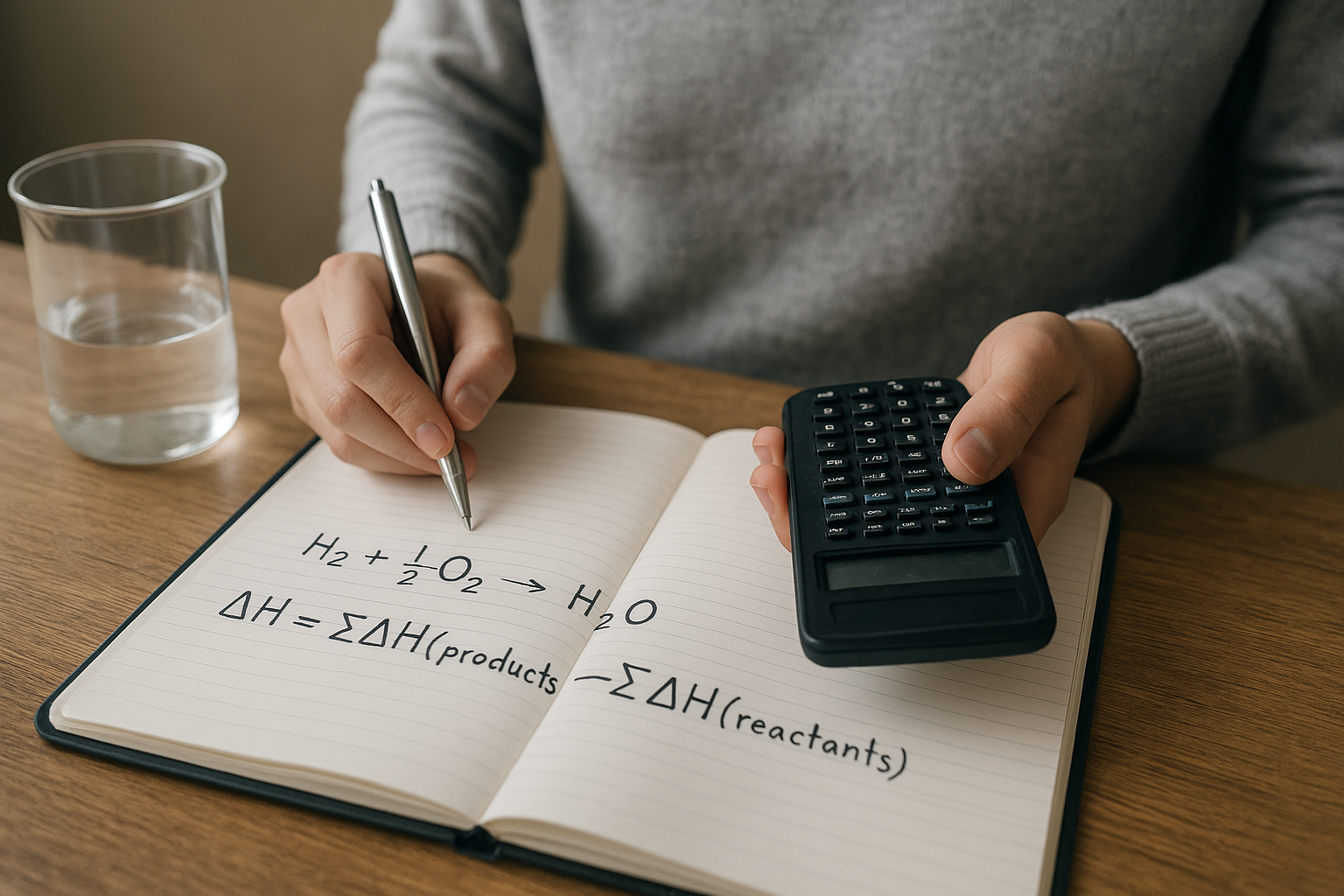
Reakcióhő számítása: Hogyan használjuk a képződéshőket a képletben?
A reakcióhő (reakcióentalpia) azt írja le, mennyi hő szabadul fel vagy nyelődik el egy kémiai reakció során állandó nyomáson. A mindennapi kémiai gondolkodásban ez az egyik legfontosabb mennyiség, mert közvetlenül kapcsolódik a reakció „energetikájához”.
Fizikában azért kulcsfontosságú, mert a termodinamika egyik alapfogalma: az entalpia-változás segítségével megérthetjük, mi hajtja a folyamatokat, hogyan oszlik meg az energia, és hogyan kapcsolódik a hőcsere a munkavégzéshez. Ez különösen fontos, ha reakciók energetikáját kell mérnöki pontossággal számolni.
A technológiában és a hétköznapokban mindenütt ott van: üzemanyagok égése, fűtés, hőtermelő és hőelvonó kémiai folyamatok, akkumulátorok, ipari ammóniagyártás, cementgyártás, sőt még az élelmiszeripari fermentációk energetikája is ide tartozik.
Tartalomjegyzék
- Mit jelent a reakcióhő, és miért fontos?
- Képződéshő fogalma: alapok és definíciók
- Standard állapotok: mikor érvényesek az adatok?
- A Hess-törvény lényege reakcióhő számításban
- A képlet: ΔHreakció a képződéshőkből számolva
- Termékek és kiindulási anyagok helyes kezelése
- Előjelek szerepe: mikor vonunk ki, mikor adunk?
- Táblázatok használata: hol találjuk a ΔHf értékeket?
- Lépésről lépésre példa egy egyszerű reakcióra
- Összetettebb egyenletek: együtthatók és szorzás
- Gyakori hibák a reakcióhő kiszámításakor
- Ellenőrzés és becslés: reális-e a kapott ΔH?
Mit jelent a reakcióhő, és miért fontos?
A reakcióhő a kémiai reakció során bekövetkező entalpiaváltozás (ΔH). Ha a reakció állandó nyomáson zajlik (laborban gyakori), akkor a mért hőcsere jó közelítéssel egyenlő az entalpiaváltozással. Ezért a „reakcióhő” kifejezést sokszor a ΔH szinonimájaként használjuk.
Gyakorlati jelentősége óriási: megmondja, hogy a reakció exoterm (hőt termel) vagy endoterm (hőt nyel el), és mennyire. Például az égési reakciók nagy negatív ΔH-ja teszi lehetővé a motorok, kazánok működését; míg egy endoterm reakció (például néhány só oldódása) hűtőtasakok alapja lehet.
Képződéshő fogalma: alapok és definíciók
A (standard) képződéshő, ΔHf°, annak az entalpiaváltozása, amikor 1 mol vegyület elemeiből képződik, mégpedig az elemek standard állapotában. Ez egy táblázatosan közölt „alapadat”, amiből rengeteg reakció ΔH-ja kiszámítható mérés nélkül.
Példa: a víz standard képződéshője azt írja le, milyen entalpiaváltozás tartozik ahhoz, hogy hidrogénből és oxigénből víz keletkezzen standard körülmények között. Fontos szabály: az elemek standard állapotban vett képződéshője definíció szerint 0 (pl. O₂(g), N₂(g), grafit C(s), Fe(s) stb.).
Standard állapotok: mikor érvényesek az adatok?
A táblázatokban szereplő ΔHf° értékek csak akkor használhatók közvetlenül, ha a reakcióban szereplő anyagok standard állapotban vannak. Ez jellemzően 1 bar nyomást jelent, és egy megadott hőmérsékletet (leggyakrabban 298,15 K, azaz 25 °C), valamint a tiszta anyagok referencia fázisát (szilárd, folyadék, gáz, oldat).
A gyakorlatban sok reakció nem pontosan ilyen körülmények között zajlik, mégis gyakran használjuk az adatokat, mert jó közelítést adnak. Haladó szinten viszont számolni kell azzal, hogy a ΔH hőmérsékletfüggő (hőkapacitások), illetve fázisfüggő (pl. H₂O(l) és H₂O(g) képződéshője nem ugyanaz). Mindig ellenőrizd a táblázatban a fázisjelölést.
A Hess-törvény lényege reakcióhő számításban
A Hess-törvény lényege: az entalpia állapotfüggvény, ezért a reakcióentalpia csak a kezdeti és végállapotoktól függ, nem az úttól. Magyarán: ha egy reakciót több lépésben is le lehet írni, a teljes ΔH ugyanaz lesz, mint az összes részlépés ΔH-jának összege.
Ez az elv teszi lehetővé, hogy a képződéshőket „építőkockaként” használjuk. A táblázatos ΔHf° értékek lényegében olyan referencia-lépések entalpiái, amelyekből bármilyen reakció ΔH-ja összeállítható. Nem kell kalorimetriát végezni minden egyes reakcióra, elég a megbízható adatbázis és a helyes egyenletkezelés.
A képlet: ΔHreakció a képződéshőkből számolva
A számítás alapképlete a „termékek mínusz kiindulási anyagok” elv. A reakcióentalpiát úgy kapod meg, hogy összeadod a termékek standard képződéshőit a sztöchiometriai együtthatóikkal szorozva, majd kivonod a kiindulási anyagok megfelelő összegét. Ez nem trükk: a Hess-törvény közvetlen következménye.
Itt különösen fontos, hogy a reakcióegyenlet helyesen legyen kiegyenlítve, és minden anyagnál a megfelelő fázis szerepeljen. Ugyanaz a kémiai összegképlet (pl. H₂O) más fázisban más ΔHf°-t jelent, és ez tipikusan több tíz kJ/mol eltérést okoz—ami már mérnöki számításban nagyon nagy hiba.
ΔH°reakció = Σ νᵢ ΔHf° termék,i − Σ νⱼ ΔHf° kiindulás,j
Termékek és kiindulási anyagok helyes kezelése
A képlet használata valójában könyvelési feladat: mindent rendszerezetten kell összesíteni. A termékoldalon (jobb oldal) szereplő anyagok ΔHf° értékeit összeadod, mindegyiket megszorzod a saját anyagmennyiségi együtthatójával (ν). Ugyanezt megcsinálod a bal oldallal is, és a kettő különbsége adja a reakcióentalpiát.
A „helyes kezelés” legfontosabb része, hogy a reakcióegyenletben szereplő együtthatók nem díszek. Ha például 2 mol víz keletkezik, akkor a víz ΔHf°-ját 2-vel kell szorozni. Haladó feladatokban gyakori, hogy törtszámokkal kényelmesebb kiegyenlíteni; ez teljesen rendben van, de következetesen kell vinni az egész számítást.
Előjelek szerepe: mikor vonunk ki, mikor adunk?
A legtöbb hiba itt történik. A képletben nem az előjelek „mágikusak”, hanem a logika: termékösszeg − kiindulási összeg. A ΔHf° értékek önmagukban lehetnek negatívak vagy pozitívak; te viszont először mindig összegeket képezel, és csak utána vonsz ki.
Érdemes fejben ellenőrizni: ha a termékek entalpiája (a képződéshők alapján) „alacsonyabb”, mint a kiindulási anyagoké, akkor ΔH°reakció negatív lesz, tehát exoterm. Ha fordítva, akkor pozitív, endoterm. A negatív ΔH nem „rossz”, hanem hőfelszabadulást jelent.
Táblázatok használata: hol találjuk a ΔHf értékeket?
ΔHf° adatokat megbízható kémiakönyvekben, adatlapokon és adatbázisokban találsz. Tipikus források: egyetemi fizikai kémia táblázatok, NIST Chemistry WebBook, CRC Handbook, illetve tankönyvi függelékek. Kezdőként is érdemes megtanulni, hogy mindig nézd a hőmérsékletet és a fázist.
A táblázatokban sokszor szerepel még ΔGf° és S° is; ezek más célra valók (szabadentalpia, entrópia), de ugyanaz a logika (termékek − kiindulási anyagok) gyakran előjön. Reakcióhő számításnál kifejezetten a ΔHf° oszlop kell, és az anyag pontos jelölése (pl. CO₂(g) vs CO₂(aq)).
| Anyag | Fázis | ΔHf° tipikus előjel | Megjegyzés |
|---|---|---|---|
| O₂ | g | 0 | elem standard állapotban |
| C | grafit, s | 0 | a gyémánt nem standard referencia |
| H₂O | l | negatív | erősen stabil, exoterm képződés |
| H₂O | g | negatív | eltér a folyadéktól párolgáshő miatt |
Lépésről lépésre példa egy egyszerű reakcióra
Vegyük a metán égését, ami gyakorlati szempontból is ismerős (földgáz). Reakció (kiegyenlítve, standard állapotokra figyelve): CH₄(g) + 2 O₂(g) → CO₂(g) + 2 H₂O(l). Most csak a módszer a lényeg: a termékek és kiindulási anyagok ΔHf°-ját kell összerakni.
Tegyük fel (szokásos 298 K körüli táblázatból), hogy: ΔHf°[CH₄(g)] negatív, ΔHf°[CO₂(g)] negatív, ΔHf°[H₂O(l)] erősen negatív, ΔHf°[O₂(g)] = 0. Ezeket behelyettesítve kijön egy nagy negatív szám, ami pontosan azt fejezi ki, hogy az égés sok hőt termel.
ΔH°reakció = ΔHf°[CO₂(g)] + 2ΔHf°[H₂O(l)] − ΔHf°[CH₄(g)] − 2ΔHf°[O₂(g)]
ΔHf°[O₂(g)] = 0
ΔH°reakció = ΔHf°[CO₂(g)] + 2ΔHf°[H₂O(l)] − ΔHf°[CH₄(g)]
Összetettebb egyenletek: együtthatók és szorzás
Összetettebb reakcióknál (pl. több termék, több lépés, ionok, hidratált formák) a módszer ugyanaz, csak több tag lesz. A legfontosabb készség: előbb mindig egyenlítsd ki a reakcióegyenletet, és csak utána számolj. Ha menet közben változtatod az együtthatókat, szinte biztos, hogy elrontod a végösszeget.
Például etanol égése: C₂H₅OH(l) + 3 O₂(g) → 2 CO₂(g) + 3 H₂O(l). Itt már látszik, hogy minden ΔHf°-t a megfelelő szorzóval kell venni (2 és 3), és az oxigén itt is 0 marad standard állapotban. Haladóként érdemes megjegyezni: ha a víz gőz fázisban keletkezik (H₂O(g)), akkor a reakcióentalpia kevésbé negatív lesz, mert a párolgás „energiát visz el”.
ΔH°reakció = 2ΔHf°[CO₂(g)] + 3ΔHf°[H₂O(l)] − ΔHf°[C₂H₅OH(l)] − 3ΔHf°[O₂(g)]
ΔHf°[O₂(g)] = 0
ΔH°reakció = 2ΔHf°[CO₂(g)] + 3ΔHf°[H₂O(l)] − ΔHf°[C₂H₅OH(l)]
Gyakori hibák a reakcióhő kiszámításakor
Az egyik leggyakoribb hiba a fázisok elnézése. Ha például H₂O(l) helyett H₂O(g) értékkel számolsz, a hiba nagyságrendileg a párolgáshővel összemérhető, ami víznél jelentős. Ugyanígy: grafit vs gyémánt, vagy különböző allotrópok, hidratációs állapotok—mind eltérő ΔHf°-t jelentenek.
A másik tipikus hiba a sztöchiometriai szorzók elfelejtése, illetve az előjeles kivonás elrontása. Ilyenkor segít egy „könyvelős” táblázat: külön oszlop a termékeknek és kiindulási anyagoknak, külön a ν szorzónak, és a végén két összeg és egy kivonás. Ha rendszerezetten írsz, a hibák nagy része eltűnik.
| Hibatípus | Mi történik? | Következmény | Javítás |
|---|---|---|---|
| Fázis rossz | H₂O(l) helyett H₂O(g) | nagy eltérés kJ/mol-ban | mindig ellenőrizd a (s)(l)(g)(aq) jelölést |
| Együttható kimarad | 2 helyett 1-szeres szorzás | arányos hiba | reagensek elé írt ν mindig szoroz |
| O₂ képződéshőjét nem 0-nak veszik | „kitalált” adat | teljes eredmény torzul | elem standard állapotban: ΔHf° = 0 |
| Nem kiegyenlített egyenlet | rossz anyagmérleg | értelmetlen ΔH | először egyenlíts, csak utána számolj |
Ellenőrzés és becslés: reális-e a kapott ΔH?
Ellenőrzéshez először nézd meg az előjelet. Égésnél (szerves anyag + O₂ → CO₂ + H₂O) szinte mindig ΔH°reakció negatív és elég nagy abszolút értékű. Ha pozitív jön ki, akkor valószínű előjel- vagy fázishiba történt. Endoterm példák (pl. CaCO₃(s) → CaO(s) + CO₂(g)) esetén a pozitív ΔH teljesen reális.
Második lépésként becsüld meg a nagyságrendet. Egy „átlagos” kovalens kötéseket átrendező reakció entalpiája gyakran néhány tíz vagy pár száz kJ/mol tartományban van. Ha ezer kJ/mol nagyságrendű értéket kapsz egy egyszerű oldódásra, az gyanús. Haladó ellenőrzés: hasonlítsd össze ismert irodalmi égéshőkkel vagy hasonló reakciók adataival.
| Ellenőrzési módszer | Kezdő szinten hasznos? | Haladó szinten hasznos? | Mire jó? |
|---|---|---|---|
| Előjel-logika (égés exoterm) | igen | igen | gyors hibaszűrés |
| Nagyságrend-becslés | igen | igen | irreális értékek kiszúrása |
| Fázisok ellenőrzése | igen | igen | tipikus nagy hibaforrás |
| Irodalmi összevetés | néha | igen | validálás, publikálható pontosság |
Kémiai definíció
A reakcióhő (reakcióentalpia) ΔHreakció az a mennyiség, amely megadja a rendszer entalpiájának változását a reakció során állandó nyomáson. Standard esetben ΔH°reakció-ról beszélünk, ha minden résztvevő standard állapotban van.
Rövid magyarázat példával: ha a reakció exoterm, akkor a rendszer entalpiája csökken, ezért ΔH negatív. Például sok égési reakcióban a termékek (CO₂, H₂O) stabilabbak, így alacsonyabb entalpiájúak, mint a kiindulási anyagok.
Jellemzők, szimbólumok / jelölések
A reakcióhő számításához tipikusan az alábbi mennyiségekkel találkozol. Ezek skalár mennyiségek (nincs irányuk), előjelük viszont fizikai jelentésű.
- ΔH°reakció: standard reakcióentalpia, kJ/mol reakció (a megírt egyenlet szerinti „1 mol reakcióra”).
- ΔHf°: standard képződéshő (entalpia), kJ/mol anyag.
- ν: sztöchiometriai együttható (dimenziótlan).
- Σ: összegzés jel, termékekre és kiindulási anyagokra külön.
Előjel-konvenció: ΔH < 0 exoterm, ΔH > 0 endoterm. Fontos: az egyenlet megfordításakor ΔH előjelet vált; az egyenlet minden együtthatójának n-szeresére szorzásakor ΔH is n-szeresére változik.
ΔH°reakció = Σ νᵢ ΔHf° termék,i − Σ νⱼ ΔHf° kiindulás,j
Típusok
A reakcióhőt több szempont szerint szokás osztályozni. Kezdőként elég az exoterm–endoterm felosztás, haladóként pedig fontos a „milyen reakciótípushoz” tartozó entalpiák ismerete.
Gyakori típusok:
- Égéshő: anyag teljes égése O₂-ben (általában nagy negatív ΔH).
- Oldáshő: oldódáskor fellépő entalpiaváltozás (lehet pozitív vagy negatív).
- Semlegesítési hő: sav-bázis reakciók entalpiája (vizes közegben gyakran jellegzetes értékek).
- Bomlási entalpia: vegyület elemeire vagy egyszerűbb anyagokra bomlása (gyakran a képződés „ellentéte”).
Haladó megjegyzés: ugyanazt a folyamatot leírhatod belsőenergia-változással (ΔU) is, de laboratóriumi, nyitott edényes körülmények között a gyakorlatban a ΔH a kényelmesen kezelhető mennyiség.
Képletek és számítások
A fő képlet a képződéshőkből számolt reakcióentalpia. Ez azt fejezi ki, hogy a reakció „összentalpiája” a termékek képződésének entalpiájából és a kiindulási anyagok képződésének entalpiájából állítható elő Hess-törvény alapján.
Minden jel a képletben:
- ν: a kiegyenlített reakcióegyenlet együtthatója
- ΔHf°: standard képződéshő adott anyagra és fázisra
- Σ: összeadás minden termékre, illetve minden kiindulásra
ΔH°reakció = Σ νᵢ ΔHf° termék,i − Σ νⱼ ΔHf° kiindulás,j
Egyszerű „komponens” felírás (két termék, két kiindulás példáján):
ΔH°reakció = ν₁ΔHf°₁ + ν₂ΔHf°₂ − ν₃ΔHf°₃ − ν₄ΔHf°₄
SI mértékegységek és átváltások
Az entalpia Joule-ban mérhető, de kémiában szinte mindig kJ/mol formában találkozol vele. A „mol” itt azt jelenti, hogy az érték 1 mol anyagra (képződéshő) vagy a reakcióegyenlet szerinti 1 mol reakcióra (reakcióentalpia) vonatkozik.
Gyakori átváltások és előtagok:
- 1 kJ = 1000 J
- 1 MJ = 10⁶ J
- 1 J = 1 N·m
- 1 kJ/mol = 1000 J/mol
- m = milli = 10⁻³, µ = mikro = 10⁻⁶, k = kilo = 10³, M = mega = 10⁶
| Mennyiség | Tipikus egység | SI alap | Megjegyzés |
|---|---|---|---|
| ΔHf° | kJ/mol | J/mol | táblázatokban leggyakrabban kJ/mol |
| ΔH°reakció | kJ/mol | J/mol | „mol reakcióra” értendő |
| T | K | K | standard gyakran 298,15 K |
| p | bar vagy Pa | Pa | standard állapot tipikusan 1 bar |
GYIK – 10 kérdés és válasz
-
Mi a különbség a reakcióhő és a képződéshő között?
A reakcióhő (ΔHreakció) egy konkrét reakció entalpiaváltozása, a képződéshő (ΔHf°) pedig egy anyag elemeiből való képződésének standard entalpiája. -
Miért 0 az elemek ΔHf° értéke standard állapotban?
Mert ez definíció: a referenciaállapotot így rögzítjük, hogy minden más érték ehhez legyen viszonyítva. -
Mit jelent a „standard” jel (°)?
Azt, hogy az adat standard állapotokra (tipikusan 1 bar, 298,15 K, megfelelő referenciafázis) vonatkozik. -
Ha megfordítom a reakciót, mi történik ΔH-val?
ΔH előjelet vált: ami exoterm volt, endoterm lesz ugyanakkora abszolút értékkel. -
Ha az egyenletet 2-vel szorzom, ΔH is duplázódik?
Igen. ΔH extenzív a reakció „mértékére”: az együtthatók skálázásával arányosan változik. -
Miért kell a fázisokra figyelni?
Mert az entalpia függ a halmazállapottól; H₂O(l) és H₂O(g) képződéshője különbözik. -
Mi a leggyakoribb előjelhiba?
A „termékek − kiindulási anyagok” elv elcserélése, vagy az, hogy a negatív ΔHf° értékek kivonásakor rosszul kezelik a mínusz jelet. -
Használhatom a képletet oldott (aq) anyagokra is?
Igen, ha a táblázat ad ΔHf° értéket az adott (aq) formára, és a reakció is ehhez illeszkedik. -
Mit jelent az, hogy kJ/mol reakció?
Azt, hogy a megírt, kiegyenlített reakcióegyenlet egyszeri „lefutására” (a sztöchiometriai mennyiségekre) vonatkozik. -
Honnan tudom gyorsan, hogy reális-e az eredmény?
Nézd az előjelet (égésnél negatív), a nagyságrendet (tízes–százas kJ/mol gyakori), és ellenőrizd a fázisokat, együtthatókat, valamint hogy O₂, N₂, grafit stb. ΔHf°-ja 0-e.

